

Executive Summary
Augment Therapeutics is an early-stage pharmaceutical research and development company, and its mission is to “Leveraging Off-Patent Medicines” and strives to invent “Enhanced” versions of many widely used existing generic drugs that suffer from significant drawbacks or limitations by using our proprietary novel linker/prodrug technologies. In the process, we have invented the below mentioned “Enhanced” versions of widely used existing drugs such as naproxen, aspirin, and irinotecan. We strongly believe that these “Enhanced Medicines or E-Medicines”, due to their competitively advantageous properties over their respective parent drugs, are ideal candidates for development through USFDA’s efficient 505(b)(2) regulatory route, which is the least expensive and shortest route to drug development and approval without the need for conducting very expensive and time-consuming phase II and Phase III studies.

THE COMPANY AND TEAM
Augment Therapeutics is headquartered in the city of Foothill Ranch, California, USA and in Hyderabad, Telangana State, India. The co-founders/shareholders of the US Entity are Mr. Apparao Satyam, PhD (US Citizen), Mr. Somesh Sharma, PhD (US Citizen), Mr. Larry Kauvar, PhD (US Citizen) and Mr. Vidyasagar Vuligonda, PhD (US Citizen). The co-founders/shareholders of the Indian Entity are Mrs. Veeraveni Baddireddi (Indian citizen & Co-founder) and Mr. Apparao Satyam, PhD (US citizen & Co-founder), A PhD Pharmacologist (Indian Citizen & Shareholder) and A Clinical Research Physician, MD (Indian Citizen & Shareholder).
Augment Scientific Team – Responsible of preclinical and clinical research work

Dr. Apparao Satyam, PhD (US Citizen)
Co-founder and Chief Scientific Officer at Augment Therapeutics
30+ Years of drug discovery experience in both Indian and US Pharma industry
PhD Pharmacologist (Indian Citizen)
18+ Years of experience in preclinical research
Clinical Research Physician, MD
(Indian Citizen)
18+ Years of experience in clinical research
Augment Scientific Team – Responsible for dealings with US FDA on regulatory issues
Dr. Somesh Sharma, PhD (US citizen)
35+ Years of drug discovery experience in both Indian and US Pharma industry

Dr. Larry Kauvar, PhD (US citizen)
35+ Years of experience of drug discovery experience in US Pharma industry

Dr. Vidyasagar Vuligonda (US citizen)
30 years of experience in small molecule drug design and product development
A Regulatory Consultant, who is an expert in 505(b)(2) NDA submissions – to be hired.
Administrative Team – Responsible for managing Admin & Finance Departments

Mrs. Veeraveni Baddireddi, MMS (Finance) (Indian Citizen Woman)
Co-founder & Managing Director at Augment Therapeutics Pvt Ltd (INDIA)
Auditor: Mrs. Radhika Vunnam, Chartered Accountant, Hyderabad, India
Project 1. Development of NO-Naproxen as “Enhanced” version of widely used pain AND ARTHRITIS medicine naproxen through 505(b)(2) REGULATORY route:
Generic naproxen, a non-steroidal anti-inflammatory drug (NSAID), is widely used for the treatment of pain and other inflammatory conditions such as rheumatoid arthritis, osteoarthritis, ankylosing spondylitis, tendinitis, bursitis, gout, menstrual cramps, etc. However, like most other NSAIDs, long-term use of naproxen is associated with severe gastrointestinal (GI) lesions, ulcers, and bleeding. Additionally, recent studies have shown an increased risk of major cardiovascular events such as rise in blood pressure within 30 days of an NSAID use. To reduce the NSAID-induced GI complications, a few approaches such as co-medication with acid suppressants such as proton pump inhibitors (PPIs) and prostaglandin analogues are currently used. However, long term use of PPIs has shown many side effects. In the mid 90’, COX-2 selective inhibitors were introduced as gastro-protective NSAIDs and they became very successful within a short period of time. However, most of these COX-2 inhibitors were withdrawn from market due to their inherent cardiovascular risk. This has created a real unmet medical need for safer versions of NSAIDs which do not cause the said GI complications. Recently, a new class of nitric oxide (NO) releasing prodrugs of NSAIDs or simply NO-NSAIDs, are being studied as potentially gastro-protective NSAIDs. These NO-NSAIDs mostly retain the anti-inflammatory properties of parent NSAID, but they also exhibit significantly reduced GI toxicity, which is attributable to the beneficial effects of NO released from these compounds.
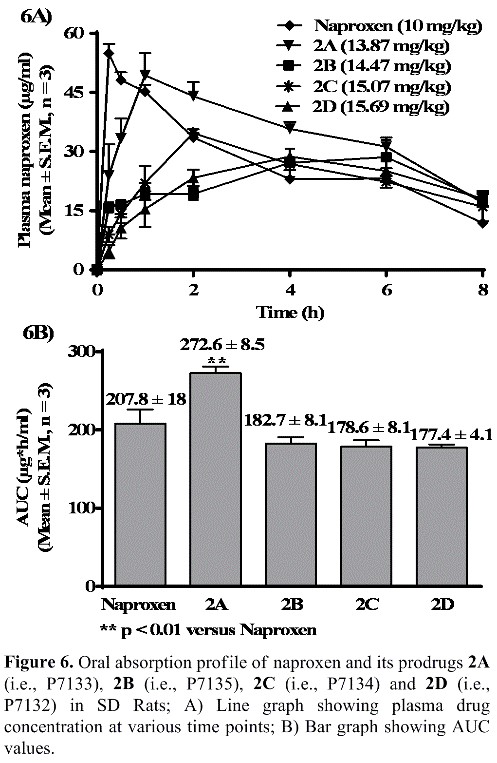
We have now discovered a novel NO-Naproxen prodrug (2A/P7133/AT-002), which is aptly named as “Enhanced Naproxen” or “E-Naproxen”, by using a novel platform linker technology and our E-Naproxen:
- Exhibited statistically significant superior bioavailability than naproxen in rats [See Figure 6 showing comparison of bioavailability data among naproxen and its prodrugs 2A (P7133/AT-002), 2B (P7135), 2C (P7134) and 2D (P7132)];
- Protected experimental rats from NSAID-induced gastric damage, which could be due to the beneficial effects of NO released from this NO-NSAID (See Figures S2 and 9).
- May show anti-hypertensive potential due to release of NO, which is a known vasodilator and regulator of blood pressure.
Thus, as shown in Figure 6, our E-Naproxen (2A/AT-002) has exhibited statistically significant increase in bioavailability (AUC: 272.60 ± 8.50 mg*h/ml, **p <0.01 vs naproxen) over its parent drug naproxen [AUC: 207.80 ± 18.20 mg*h/ ml, **p <0.01 vs NO-Naproxen (2A/AT-002)]. To our knowledge, this is the first report of a naproxen prodrug having a statistically significant increase in bioavailability than its parent drugs naproxen.
Interestingly, as shown in Figure 6, our E-Naproxen (2A/AT-002) has exhibited a more controlled or sustained release of naproxen and maintained a higher plasma drug concentration over a longer period (>30 micrograms/ml plasma drug concentration up to 6 hrs. duration) when compared to that of naproxen at equimolar doses. Our E-Naproxen is therefore expected to offer better pain relief over a longer period than the parent drug naproxen.
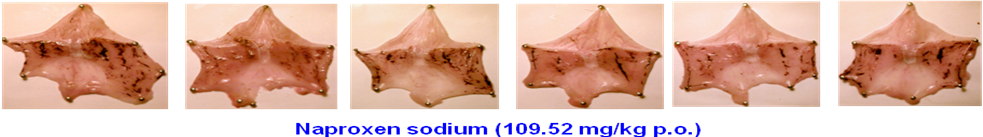
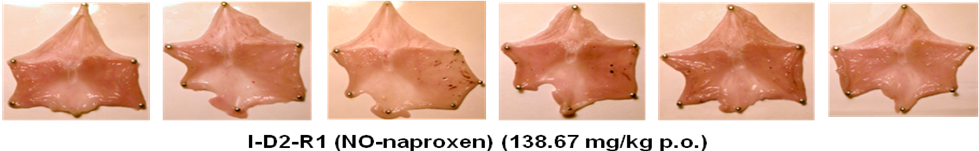
Figure S2. Images of rat stomachs showing gastric lesion and ulcer induction/sparing following acute oral administration of naproxen sodium (109.52 mg/kg, which is equimolar to 100 mg/kg dose of naproxen) and its promising NO-naproxen prodrug 2A (I-D2-R1 or P7133 or AT-002) at 138.67 mg/kg, which is a dose equimolar to 100 mg/kg dose of naproxen in rats.
Strong Possibility for Drug Development and Approval through 505(b)(2) NDA route: We strongly believe that our E-Naproxen (P7133/A2/AT-002), with its above-mentioned competitively advantageous properties over naproxen, is an ideal candidate for development through 505(b)(2) regulatory route, which is the least expensive shorter route for drug approval.
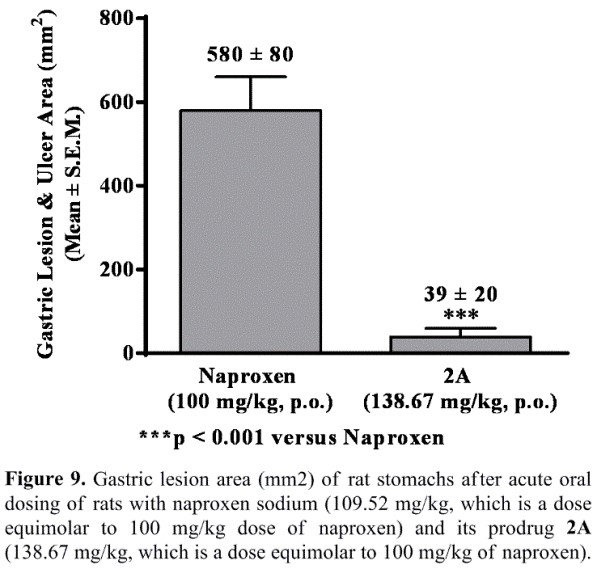
The data shown in Figures S2 and 9 clearly demonstrate that the animals treated with E-Naproxen (2A/AT-002) caused only minimal or negligible gastric lesions. However, severe hemorrhagic lesions and ulcers were developed in rats administered with parent drug naproxen at equimolar doses (i.e., Gastric lesion and ulcer area: 39 ± 20 mm3 for E-Naproxen vs 580 ± 80 mm3 for naproxen; *** p <0.001). We believe that the observed gastric-sparing effects of our promising E-Naproxen (2A/AT-002) could be attributable to gastro-protective properties of NO released from this novel NO-Naproxen.
Competitive Advantages and Market Impact: The above-mentioned positive attributes make the E-Naproxen (P7133/2A/AT-002) competitively advantageous over naproxen. We therefore believe that this E-Naproxen could represent a novel “First-in-the-Class” of “Safer NSAID” for the treatment of arthritic pain and a host of inflammatory diseases. Thus, this E-Naproxen, when it is approved, has the greatest potential to create disruption in NSAIDs market by grabbing significant market share from naproxen and other approved NSAIDs and has the potential to become a BLOCKBUSTER drug within a short period of its introduction to market.
IP/Patents: We have already obtained US, Canadian and Indian patents covering our E-Naproxen [See US 9,844,599, (Dec 19, 2017), CA 2 897 571 C (Dec 18, 2018) and IN 345054 (Aug 25, 2020)]. We have also published this work [See Bioorganic & Medicinal Chemistry Letters, 2014, 24, 5587-92].
Project 2. Discovery of a novel water-soluble prodrug of SN-38 as an “Enhanced Irinotecan” or “E-Irinotecan” or “AT-005”.
Irinotecan (Camptosar) is in the WHO-list of essential medicines and used for treating metastatic colorectal cancer [either alone or in combination with capecitabine or fluorouracil (5-FU) and leucovorin] and small cell lung cancer (with cisplatin). Irinotecan is also used in combination with folinic acid to treat pancreatic cancers. There were >$12.5B of global sales of colorectal, lung and pancreatic cancer drugs in 2021.
However, significant side effects of Irinotecan include life-threatening diarrhoea and immunosuppression due to onset of neutropenia. In fact, Irinotecan is a water-soluble prodrug of SN-38, which is a highly potent camptothecin analog. However, it was reported that irinotecan releases only 2-5% of SN-38 in vivo in humans and it is indeed a significant defect as SN-38 release from Irinotecan is dependent on limitedly available carboxylesterases (CES1 and CES2) in the liver and butyryl cholinesterase (hBChE) in plasma. To overcome this problem, we have discovered an “Enhanced Irinotecan” or “E-Irinotecan” or “AT-005”, a new water-soluble prodrug of SN-38, using a novel self-immolative linker that can be cleaved at biological pH (~7.2) to release 100% of SN-38 in vivo. Hence, our E-Irinotecan (AT-005) does not depend on liver carboxylesterases or plasma butyryl cholinesterase to release SN-38 in humans.
Although the areas under the plasma concentration-time curve (AUC) of both irinotecan and SN-38 increase proportionally to the administered dose, SN-38 levels achieved in humans are about 100-fold lower than the corresponding irinotecan levels, but these concentrations are important since SN-38 is 100- to 1,000-fold more cytotoxic than irinotecan. It was also observed that AUCs of Irinotecan and SN-38 correlate significantly with leuko-neutropenia and with the intensity of diarrhoea. However, it was observed that most tumor responses were seen at the highest doses administered in Phase I trials suggest a dose-response relation with Irinotecan. These PK-PD relationships are important for a better clinical management of Irinotecan aimed at a better control of toxicities and a better prediction of tumor response for the benefit of an individual patient.
Since our E-Irinotecan (AT-005) releases SN-38 quantitatively at biological pH and does not depend on any bio-enzymes for its conversion, it would be practical for clinical physicians to design and conduct a few clinical trials to determine an optimal dose of E-Irinotecan (AT-005) that is clinically effective but minimally toxic. One major advantage with our E-Irinotecan is that we don’t see any of the toxicities associated with the drug Irinotecan. In other words, we need to consider only the known toxicities associated with SN-38 exposure, when designing clinical trials to determine a clinically effective, but minimally toxic, dose of E-Irinotecan (AT-005).
Although both Irinotecan and E-Irinotecan are water-soluble prodrugs of SN-38 and possess a stable tert-urethane linkage, they release SN-38 to a different extent under different conditions as shown in the following in vitro and in vivo experiments:
In Vitro SN-38 release from Irinotecan & E-Irinotecan (AT-005) in human plasma
|
Time (min) |
Release of SN-38 in human Plasma |
|
|
Irinotecan |
E-Irinotecan (AT-005) |
|
|
0 |
0 |
0 |
|
15 |
0 |
100 |
|
30 |
0 |
100 |
|
120 |
0 |
100 |
|
Half-life (t1/2) |
Stable (>2hrs) |
<15 min |
In Vivo (PK) SN-38 release data for Irinotecan & E-Irinotecan (AT-005) in Mice
|
Time (min) after iv administration |
Release of SN-38 in vivo (in mice) |
|
|
Irinotecan |
E-Irinotecan (AT-005) |
|
|
2 |
~1200 ng/ml |
~2500 ng/ml |
|
5 |
~800 ng/ml |
~850 mg/ml |
|
15 |
~500 ng/ml |
~750 ng/ml |
|
30 |
~300 ng/ml |
~350 ng/ml |
|
60 |
~50 ng/ml |
~75 ng/ml |
In vitro study: As expected, our E-Irinotecan (AT-005) released 100% of SN-38 within 15 min of incubation in human plasma. However, Irinotecan did not release any amount of SN-38 even after 2 hours of incubation in human plasma.
In vivo (PK) Study: When equimolar amounts of both the prodrugs were administered to mice via iv, Irinotecan released about 1200 ng/ml of SN-38, whereas E-Irinotecan released nearly double the amount (i.e., 2500 ng/ml) of SN-38 in vivo. This result is not surprising as rodents possess higher amounts of liver carboxylesterases and humans possess only limited amount of liver carboxylesterases.
Based on the above results,
Can <10-20% dose of E-Irinotecan (AT-005) be therapeutically equivalent to the full dose of Irinotecan?
Can use of such lower doses of E-Irinotecan (AT-005) have the potential to reduce the intensity of SN-38/Irinotecan-associated major side effects such as diarrhoea and neutropenia?
We don’t know answers yet!
However, we are planning to conduct a few relevant in vitro and in vivo preclinical (and eventually clinical) studies to find answers.
Project 3. NO-Aspirin as an “Enhanced” version of the widely used wonder drug aspirin:
Aspirin has been in use as an anti-inflammatory and anti-pyretic drug for over 100 years. Recently, aspirin is also widely used for the treatment of cardiovascular diseases. However, significant number of aspirin users suffer from NSAID-induced severe gastrointestinal lesions, bleeding and ulcers. Currently, proton pump inhibitors are used in combination with aspirin to reduce NSAID-induced GI damage. However, long-term use of PPIs leads to many side effects such as osteoporotic fractures. So, there is an immediate unmet medical need for safer versions of aspirin. As a solution, we have now invented an “Enhanced Aspirin” or “E-Aspirin” or “AT-003”, which has shown comparable oral bioavailability and antiplatelet activity (i.e., TXB2 inhibition) to those of aspirin but it did not cause any significant NSAID-induced gastric lesions, bleeding, and ulcers. Thus, our E-Aspirin (AT-003) represents a First-in-Class of potentially “Safe Aspirin” for the treatment of cardiovascular disorders and is an ideal candidate for development through the least expensive 505(b)(2) regulatory route. We have already obtained US, Canadian and Indian patents covering our E-Aspirin [See US 9,844,599, (Dec 19, 2017), CA 2 897 571 C (Dec 18, 2018) and IN 345054 (Aug 25, 2020)]. We have also published this work [See Bioorganic & Medicinal Chemistry Letters, 2014, 24, 5587-92].
Exclusive Licensing of Proprietary Novel Linker Technologies: An exclusive license agreement to the technology has been secured from Piramal Enterprises, Mumbai, India. The following patents have been granted on the novel linker technology covering E-Naproxen (AT-002) and E-Aspirin (AT-003): US 9,844,599, (Dec 19, 2017), CA 2 897 571 C (Dec 18, 2018) and IN 345054 (Aug 25, 2020)].
For further Information, please contact:
Dr. Apparao Satyam, PhD,
Co-Founder & Chief Scientific Officer
Augment Therapeutics
The US Entity: 27472 Portola Parkway, Ste 205-213, Foothill Ranch, CA, USA
The Indian Entity: Villa 03, Greenspace Emerald, Puppalguda, Hyderabad-500089, India
Tel: +91-9502043658 (WhatsApp: 9502043658)
Email: apparao.satyam@augmenttherapeutics.com
Copyright © 2020. Augmenttherapeutics.com | 5